La enfermedad de Alzheimer es una demencia que cuenta ya con más de 100 años de historia. En noviembre de 1901 ingresó en el hospital de enfermedades mentales de Frankfurt una paciente de 51 años de edad llamada Auguste Deter (Augusta D), con un llamativo cuadro clínico de 5 años de evolución.
Tras comenzar con un delirio celotípico, la paciente había sufrido una rápida y progresiva pérdida de memoria acompañada de alucinaciones, desorientación en tiempo y espacio, paranoia, trastornos de la conducta y un grave trastorno del lenguaje. Fue estudiada por Alois Alzheimer, y más tarde por parte de médicos anónimos. Falleció el 8 de abril de 1906 por una septicemia, secundaria a úlceras por presión y neumonía.
El cerebro de la enferma fué enviado a Alzheimer, que procedió a su estudio histológico. El 4 de noviembre de 1906 presentó su observación anatomoclínica con la descripción de placas seniles, ovillos neurofibrilares y cambios arterioescleróticos cerebrales.
El trabajo se publicó al año siguiente con el título: «Una enfermedad grave característica de la corteza cerebral». La denominación del cuadro clínico como enfermedad de Alzheimer fue introducida por Kraepelin en la octava edición de su«Manual de psiquiatría», en 1910.
Alzheimer describió su segundo caso en 1911, fecha en la que también aparece una revisión publicada por Fuller, con un total de 13 pacientes con enfermedad de Alzheimer, con una media de edad de 50 años y una duración media de la enfermedad de 7 años.
Las lesiones histopatológicas del cerebro de Augusta D han podido ser estudiadas de nuevo y publicadas en 1998 en la revista Neurogenetics. En este trabajo no se han encontrado lesiones microscópicas vasculares, existiendo solamente placas amiloideas y ovillos neurofibrilares, lesión ésta última descrita por primera vez por Alzheimer en este cerebro.
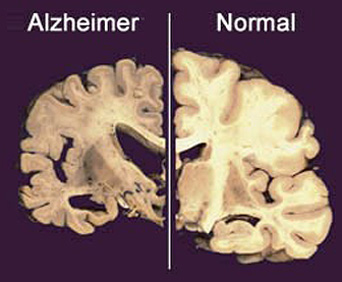
Sintomas del alzheimer.
- Pérdida de memoria que afecta a la capacidad laboral.
- Dificultad para llevar a cabo tareas familiares.
- Problemas con el lenguaje.
- Desorientación en tiempo y lugar.
- Juicio pobre o disminuido.
- Problemas con el pensamiento abstracto.
- Cosas colocadas en lugares erróneos.
- Cambios en el humor o en el comportamiento.
- Cambios en la personalidad.
- Pérdida de iniciativa.
- Repite una y otra vez las mismas cosas y hace una y otra vez las mismas preguntas, y tiene dificultades para encontrar la palabra adecuada en una conversación, utilizando parafasias y circunloquios. El rendimiento laboral es cada vez más pobre, y comienza algo más adelante a presentar ideas delirantes, culpando a familiares de esconderle o quitarle las cosas.Luego su aspecto comienza a dejar de preocuparle, y cada vez le cuesta más trabajo seguir una conversación, quedándose con frecuencia sin saber lo que iba a decir. Empieza ya a retraerse, tendiendo a dejar de salir y a abandonar sus aficiones habituales. Aparecen episodios de desorientación espacial, que inicialmente se refieren solo a los lugares menos familiares.Su percepción de la realidad es cada vez más pobre, y el cuadro evoluciona ya con rapidez hacia la demencia grave. Tiene entonces dificultades para vestirse, asearse, manejar cubiertos de manera adecuada, duerme mal, está hiperactivo (pero sin finalidad determinada), y a veces se orina en la cama.Pueden aparecer crisis epilépticas y mioclonías, y el paciente camina con lentitud, con el tronco flexionado. Orina y defeca en lugares inapropiados, apenas emite algunas palabras ininteligibles y tiene intensos trastornos del sueño y del comportamiento. Finalmente llega a no poder andar y a no comunicarse en absoluto, y fallece a causa de los procesos intercurrentes (úlceras por presión que se infectan, neumonías…).La exploración ayudará a descartar otras enfermedades sistémicas y neurológicas, y a continuación hay que confirmar los déficits cognitivos recogidos en la anamnesis, y determinar su repercusión sobre la vida social y laboral del paciente. Se exploran la orientación, concentración, memoria, lenguaje, praxias, función ejecutiva…Diágnostico.Los criterios diagnósticos de demencia de tipo Alzheimer del DSM-IV-TR (Texto revisado de la cuarta edición del manual diagnóstico y estadístico de la Asociación Americana de Psiquiatría) o los criterios actualizados en 2011 de la demencia debida a enfermedad de Alzheimer probable del National Institute on Aging y la Alzheimer's Association (NIA–AA, que sustituyen a los del NINCDS-ADRDA, National Institute of Neurologic, Communicative Disorders and Stroke - Alzheimer's Disease and Related Disorders Association) deberían ser los utilizados de manera rutinaria para la enfermedad de Alzheimer.Las siguientes pruebas complementarias deben realizarse de forma rutinaria:Determinaciones en sangre y orina: glucosa, urea, creatinina, sodio, potasio, calcio, ácido úrico, colesterol, triglicéridos, aminotransferasas, gamma-glutamiltranspeptidasa (gamma-GT), fosfatasa alcalina y albúmina, además de pruebas de función tiroidea y niveles de vitamina B12. Hemograma completo. Serología de lúes sólo si el paciente tiene factores de riesgo específicos, aunque la Sociedad Española de Neurología recomienda su realización rutinaria. Análisis elemental de orina.Pruebas de imagen: Tomografía Axial Computarizada (TAC) o Resonancia Nuclear Magnética (RNM) craneales.Punción lumbar: solamente en caso de sospecha de infección del Sistema Nervioso Central (SNC), serología de lúes positiva, hidrocefalia, edad inferior a 55 años, demencia inusual o rápidamente progresiva, inmunosupresión, sospecha de vasculitis del SNC o presencia de enfermedad metastásica.Electrocardiograma y radiología simple de tórax.Electroencefalograma: sólo si existe historia de convulsiones, pérdida de consciencia, episodios de confusión o deterioro clínico rápido.Las estrategias de medición linear o volumétrica mediante TAC o RNM no se recomiendan de manera rutinaria en la actualidad, ni la tomografía de emisión de positrones (PET). Tampoco la tomografía computarizada de emisión de fotón único (SPECT) cerebral se recomienda de manera rutinaria en el diagnóstico inicial o diferencial, ya que no ha demostrado superioridad sobre los criterios clínicos.El estudio rutinario del genotipo APOE no se recomienda actualmente en los pacientes con sospecha de enfermedad de Alzheimer, ni tampoco el de otros marcadores genéticos. No hay marcadores del líquido cefalorraquídeo (LCR) ni otros marcadores biológicos recomendados para el uso rutinario en el diagnóstico de la enfermedad de Alzheimer en estos momentos.Tratamiento.
. Tratamiento específico
Inhibidores de la colinesterasa: Se han demostrado efectos terapéuticos significativos con varios de ellos en la enfermedad de Alzheimer, indicando que son agentes mejores que el placebo de manera consistente. Sin embargo, la enfermedad sigue progresando a pesar del tratamiento, y la magnitud de la eficacia promedio es modesta (un retraso de entre 2 y 7 meses en el patrón progresivo de la evolución). Se han detectado con ellos cambios globales cognitivos, de comportamiento y funcionales.Hasta ahora no se han llevado a cabo estudios comparativos cara a cara entre estos agentes, y las diferencias principales entre ellos están en los perfiles de efectos colaterales y en su sencillez de administración. Están autorizados para su uso clínico por la Food and Drug Administration (FDA) la tacrina, el donepezilo (o donepecilo), la rivastigmina y la galantamina (escritos aquí por orden de aparición en el mercado).Memantina: Se trata de un antagonista no competitivo de los receptores NMDA (N-metil-D-aspartato), y actúa uniéndose en ellos al mismo lugar que fisiológicamente lo hace el magnesio, pero con mayor afinidad. Esto bloquea la entrada masiva de calcio que se produce en las células nerviosas cuando existe una excesiva actividad del glutamato que provoca el desplazamiento del magnesio.La memantina ha sido aprobada ya para uso clínico por la Unión Europea y por la FDA de los Estados Unidos. La indicación aprobada actualmente de manera oficial es en los casos moderados, graves y moderadamente graves (estadios 5, 6 y 7 de la GDS -Global Deterioration Scale- de Reisberg), pero ya hay estudios en marcha para conseguir su aprobación para los casos leves. Asimismo, parece ser eficaz en la demencia vascular y podría tener efectos neuroprotectores, pero todo esto necesita aún de más estudios que lo corroboren.Selegilina y vitamina E: Han demostrado eficacia en producir un cierto retraso en la evolución de la enfermedad tanto la vitamina E (a dosis de 1000 unidades internacionales (U.I.) dos veces al día) como la selegilina (a dosis de 5 mg dos veces al día), retrasando asimismo la institucionalización de los pacientes. No hay efecto aditivo entre ellos, no debiendo prescribirse simultáneamente. La vitamina E es mejor tolerada que la selegilina. Ninguno de estos dos agentes ha demostrado producir mejoras en el plano cognitivo.Prednisona y estrógenos: Pese a que varios estudios epidemiológicos sugieren efectos protectores frente al desarrollo de enfermedad de Alzheimer por parte de esteroides y estrógenos, hasta ahora los ensayos clínicos llevados a cabo para corroborarlo han arrojado resultados negativos.Antiinflamatorios no esteroideos: Un estudio prospectivo sobre 6989 personas de más de 55 años (de los que 293 desarrollaron enfermedad de Alzheimer durante un seguimiento de 6.8 años) ha demostrado una menor incidencia -estadísticamente significativa- de enfermedad de Alzheimer en los sujetos con un período de tratamiento acumulado de dos o más años con antiinflamatorios no esteroideos. Podrían tener eficacia en este sentido especialmente los antiinflamatorios no esteroideos con efecto inhibidor de la beta-secretasa.Estatinas: Varios estudios epidemiológicos retrospectivos sugieren una menor incidencia (en torno a un 70% menos de media) de enfermedad de Alzheimer entre sujetos que estaban tomando estatinas como tratamiento para controlar su colesterol. De aquí podría deducirse también un efecto de la toma de estatinas sobre la progresión de la enfermedad de Alzheimer.El mecanismo no está aún claro, aunque algunos estudios parecen indicar que las estatinas estimulan la vía de la α-secretasa para la escisión de la Proteína Precursora de Amiloide. Un estudio comunicado en la 54ª Reunión Anual de laA.A.N. (Academia Americana de Neurología) de abril de 2002, realizado sobre un total de 2581 sujetos, viene a corroborar una asociación estadísticamente significativa entre el tratamiento con estatinas y una menor incidencia de enfermedad de Alzheimer.Ginkgo biloba: los datos sobre su posible eficacia son muy limitados, y en los pocos ensayos aleatorios que se han llevado a cabo la eficacia resulta ser menor que la típica obtenida con los anticolinesterásicos.




.jpg)



